

Highlight
Understanding mating determination in Tetrahymena thermophila
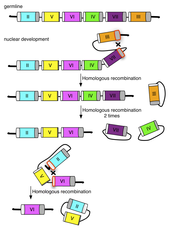
Gene excision via circle formation as a mechanism for mating type determination
Achievement/Results
When Tetrahymena thermophila mate, their progeny select 1 of 7 possible mating types. The molecular mechanism involved is unknown, but previous work has shown that the mating type of the offspring follows a nonuniform distribution that is independent of the parental mating type. IGERT Trainee Mike Lawson worked with UCSB Professor Edward Orias and postdoctoral researcher Marcella Cervantes to unravel the mystery of the molecular mechanism. Their first step was to identify the mating type genes and locate them in the genome. The result was the discovery that Tetrahymena carry copies of all mating type genes in the germline nucleus, which is transcriptionally silent during most of the cell’s life cycle but is the sole source of genetic information passed to the next generation. However, only one mating type gene is present in the transcriptionally active somatic nucleus. In the germline nucleus these genes lie next to each other, each with a highly conserved left and right region and a highly variable middle region. To put this information into a logical framework, Mike developed a simple binomial model of gene excision with one rate for removal from the left end and the second rate from the right. To test if this simple idea could explain the existing data, he optimized the two parameters in the model to fit the existing data on mating type distributions. He found a good qualitative match, however the model did not fit the exact distribution of mating types. This led to two hypotheses: first, that the assumption of removing genes in a procession from both the left and right might be incorrect and second, that the original data on mating type distributions, which is over 60 years old and relies on a phenotypic assay, might not be representative of the mechanism of gene expression.
The researchers suspected that recombination between the conserved regions was involved in selection of a single mating type gene during development, which would be consistent with the binomial model assumptions. To test this, Mike sequenced hundreds of the left and right conserved regions from the somatic nucleus of progeny, hoping to find evidence of recombination by tracking single nucleotide polymorphisms specific to each mating type. Each copy of the conserved regions required multiple partially overlapping sequencing reads to complete, which then had to be compared to a greater than 2kb reference sequence in order to identify a small number of polymorphisms. To make this data analysis tractable, Mike developed an analysis pipeline that incorporated existing software for sequence assembly from multiple reads (cap3) and sequence alignment (blastn) with his script for identifying mismatched bases between aligned sequences, thus providing a completely automated method to analyze his sequence reads in a high throughput manner. He was able to show that recombination in the conserved regions is involved in the process of selecting a single gene and was able to infer, with high statistical significance, likely sequences where the recombination occurred. This data analysis led to the hypothesis that recombination between conserved regions could form products of circular DNA (containing a mating type gene pair) that had been excised from the chromosome (see Figure 1). To test this hypothesis, Mike designed primers facing away from each other in the variable region of each mating type gene, so that polymerase chain reaction (PCR) with these primers would only produce a product if circles were formed. The experiment was successful and indicated that circles representing a single mating type are formed, a novel finding that helps to clarify the mechanism involved.
Currently, Mike and Marcella are pursuing two quantitative experiments to generate data that will enable a second generation of model development. The first experiment is to perform quantitative-PCR (qPCR) with DNA collected from a population of young progeny cells. PCR consists of multiple rounds of DNA copying; after each round the amount a specific sequence should be doubled. By fitting an exponential function to the first few data points once the product becomes detectable, it is possible to infer the number of target molecules in the starting population. The idea is to use this quantitative molecular assay to get a far more precise estimate of the distribution of mating types in the population. This may result in data that agrees more convincingly with the binomial model or it might invalidate the model’s predictions and require further explanation. The second experiment is to measure the relative rate of removal of mating type genes from the left and right by performing PCR specific to the rearranged genes, then quantifying the resultant bands using a microfulidics-based platform. Due to variability in the length of the conserved regions, circles formed by left and right cuts can have up to a one third difference in size. A difference of this amount allows the resulting band for each circle to be quantified in a relative manor. Early evidence suggests that there may be a bias between left and right side cuts that varies over time. Quantifying these relative rates will allow for refinement of the second generation model, regardless of the findings in the previous experiment.
In this work, mathematical modeling has accelerated the generation of hypotheses and high throughput data analysis has made interpretation of experimental results more efficient. The tight relationship between experiment and computation on this project has allowed for the generation of specific hypotheses that can be tested by feasible experiments, thus facilitating new discoveries. The above work has already resulted in one high profile publication, which was the cover of the journal PloS Biology, and has received recognition in broader media.
Address Goals
The fundamental contribution of this work is discovery. Identification of the location of the mating type genes and unravelling the mechanism of mating type determination provide novel insight into a process of DNA rearrangement that could have implications across all Eukaryotes. Tetrahymena thermophila has a history of being a robust model organism. For example, telomeres were first uncovered in Tetrahymena, a discovery that resulted in a Nobel prize. By uncovering the location of the genes we have provided the basis for studying homologous recombination in a highly tractable organism, which should lead to further insights by ourselves and others. In addition, we have begun detailing the mechanism involved in the rearrangement, which could have implications in cancer and immunology research.
The secondary contribution is in making our results available and accessible to the public. By publishing in an open source journal we insured that our results would be freely available to the public. In addition, we issued a press release and performed a number of interviews in the mainstream media to transmit our work to a broader audience. The result was that our work was written about in wide range of media outlets, from high level scientific journals (Nature, Science etc.) to popular science magazines (Discovery, Popular Science etc.) to general news outlets (NBC News, Huffington Post, Reddit etc.). In this way we have strived to inform the public of our work, both so they know the results of public scientific funding and to achieve a more scientifically literate public.
Last but not least, IGERT Trainee Michael Lawson had the opportunity to obtain first-hand experience in both experiment and modeling, and the synergistic feedback between the two.
